RMCProfile – I
Introduction
In
this and subsequent tutorials, we will show how to use GSAS-II in conjunction with
RMCProfile (“RMCProfile: Reverse Monte Carlo for polycrystalline materials”,
M.G. Tucker, D.A. Keen, M.T. Dove, A.L. Goodwin and Q. Hui, (2007) Jour. Phys.:
Cond. Matter, 19, 335218. doi: https://doi.org/10.1088/0953-8984/19/33/335218)
to fit both powder diffraction data and a pair distribution function (PDF)
created from it with a “big box” disordered model.
Before
getting started you must obtain the most recent Version 6 of the RMCProfile
executable (at this writing this is RMCProfile V6.7.9 although V6.7.6 will also
work, but not any earlier version). Obtain this from www.rmcprofile.org by following the
prompts from Downloads. This version is available for Windows, Mac OSX and
Linux. The download will be a zip file; save it somewhere convenient. Pull from
it only the RMCProfile_package/exe/rmcprofile.exe and the two files in RMCProfile_package/exe/cuda_lib (if present) and place them
in the GSASII main directory or in a new subdirectory (e.g. GSASII/RMCProfile); no other files are needed
although you may wish to also retrieve the contents of the tutorial
subdirectory. It contains rmcprofilemanual.pdf and rmcprofile_tutorial.pdf which may be of interest
since the GSAS-II/RMCProfile tutorials are based on the latter.
The
process for using RMCProfile begins with a normal Rietveld refinement of the
average structure. For the kind of disordered materials of interest here, this
may give bond lengths that are frequently too short for the atoms involved and
sometimes extreme apparent thermal motion for some of the atoms. These effects
arise because the average structure places these atoms too close to one
another; a more localized view would show coordinated atom displacements or
reorientation of groups of atoms to avoid the close contacts. RMCProfile is
then used to characterize these local displacements by fitting to the
diffraction data and its Fourier transform as a PDF.
The
material used in this tutorial is the same as described in the RMCProfile
tutorial Exercises 1-3, sulfur hexafluoride (SF6). The average
structure of SF6 is a molecular crystal at low temperatures with the
SF6 octahedra arranged in a body centered cubic lattice with the S-F
bonds aligned with the crystal axes. The structural frustration induced by
close F-F contacts forces the octahedra to locally rotate in different
directions and this disorder gives rise to considerable diffuse scattering. The
data used here was obtained at 190K on the GEM diffractometer at ISIS,
Rutherford-Appleton Laboratory, Harwell Campus, UK.
If you have not done so already, start GSAS-II.
Rietveld Refinement of Sulfur hexafluoride (SF6):
Step 1: Read in the data file
1. Use
the Import/Powder Data/from GSAS powder data file menu item
to read the data file into the current GSAS-II project. This read option is set
to read any of the powder data formats defined for GSAS (angles in
centidegrees, TOF in µsec). Other submenu items will read the cif format or the xye
format (angles in degrees) used by topas, etc.
In those cases, you would change the file type to cif
format or the xye format to see them.
Because you used the Help/Download tutorial menu entry to open this page and
downloaded the exercise files (recommended), then the RMCProfile-I/data/...
entry will bring you to the location where the files have been downloaded. (It is
also possible to download them manually from https://advancedphotonsource.github.io/GSAS-II-tutorials/RMCProfile-I/data/.
In this case you will need to navigate to the download location manually.)
For this tutorial you should see the data file in the file
browser, but if extensions on data files are not the expected ones, you may
need to change the file type to All files (*.*) to
find the desired file.
2. Select the sf6_190gsas.dat data file in the first dialog and press Open. There will be a Dialog box asking Is this the file you want? Press Yes button to proceed. The data file contain 3 banks of data

Select
only the third one (BANK 3) and press OK. Next will be a file selection dialog for the instrument
parameter file; only one is shown. Select it; the pattern will be read in and displayed

There
is high background and considerable diffuse scattering at low TOF (high Q)
arising from the molecular SF6 disorder.
Step 2. Set limits
Because
the Rietveld refinement will not make use of the diffuse scattering we want to
set the lower limit to just below the first visible Bragg peak; go the Limits entry in the GSAS-II data
tree.

Change
the 1st entry to 7000; the plot will change by moving the green dashed line. The
upper limit is fine.
Step 3. Import SF6
phase
The average structure of SF6 is very simple. Space group Im3m (or Im-3m; same thing), a=5.887 with S at 0,0,0 and F at 0.251,0,0. However, we will save time by importing it from an old gsas experiment file. Use the Import/Phase/from GSAS .EXP file menu item to read the phase information for SF6 into the current GSAS-II project. Other submenu items will read phase information in other formats. Because you used the Help/Download tutorial menu entry to open this page and downloaded the exercise files (recommended), then the RMCProfile-I/data/... entry will bring you to the location where the files have been downloaded. (It is also possible to download them manually from https://advancedphotonsource.github.io/GSAS-II-tutorials/RMCProfile-I/data/. In this case you will need to navigate to the download location manually.) Select the SF6_190K.EXP file (only one there) There will be a Dialog box asking Is this the file you want? Press Yes button to proceed. You will get the opportunity to change the phase name next (I left it alone; NB: because of restrictions in RMCProfile it is important that the phase name not have any spaces); press OK to continue. Next is the histogram selection window; this connects the phase to the data so it can be used in subsequent calculations.
Select the histogram (or press Set All) and press OK. The General tab for the phase is shown next
Step 4. Set the
background
Because
there is considerable diffuse scattering, the default background will be
insufficient for reasonable fitting. Select the Background item from the GSAS-II data
tree

Be
sure that “chebyschev-1” is used for the background function; RMCProfile only
recognizes that form to compute the background for the diffraction pattern.
Select 15 for the number of terms.
Step 5. Rietveld
refinement
We
are now ready for the first Rietveld refinement; select Calculate/Refine from the main GSAS-II menu.
Before it starts a file selection dialog will appear; select a file name (no
extension). I chose “SF6_190K”; do not use the phase name for this purpose because RMCProfile
will use that as the root for the many files it creates. Also, you should
create a new directory for this exercise while in this dialog box; it will be
rapidly filled up with RMCProfile files which can lead to considerable
confusion if mixed in with other files. For Windows after navigating to a
suitable location, a new directory can be made by a right mouse click and
selecting “New/Folder” from the popup menu. It will appear with the name “New
Folder”; change the name and select it (it will be empty). Other operating
systems will have similar methods. Finally press OK to save the GSAS-II project
(SF6_190K.gpx) and start the refinement. It will quickly converge to give a
quite low Rwp (~2.2%) mostly because of the very high background
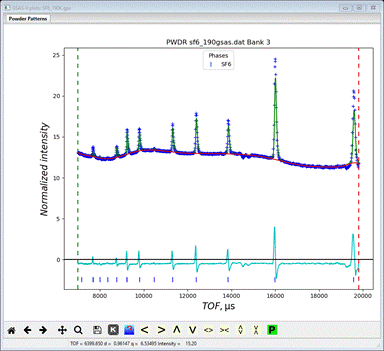
It
is evident that the lattice parameter needs adjusting. Go to the General tab for the SF6 phase and
select Refine
unit cell;
repeat Calculate/Refine. The Rwp will improve
(~0.76%) giving
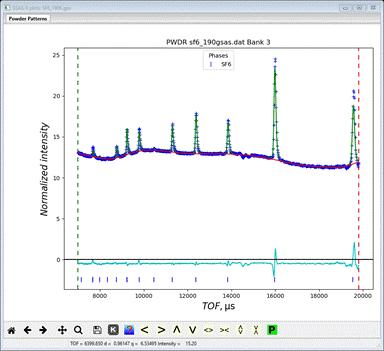
To
finish up add the following parameters:
SF6/Atoms: XU for both atoms (GSAS-II recognizes that the S atom is fixed
in position); the F atom should have anisotropic thermal displacements.
SF6/Data: refine microstrain and set size to 10.
PWDR/Instrument Parameters: refine beta-0, beta-1, sig-0, sig-1 and sig-2 (normally not necessary, but
the available GEM instrument parameter file isn’t current). In any event, the
coefficients difB, beta-q and sig-q must be zero since RMCProfile V6.7.6 Beta Serial does not
know how to use them in computing a powder profile.
Do Calculate/Refine from the main menu; the fit
will quickly improve a bit more to Rwp ~ 0.56%.
Step 6. Draw
structure
Select
the Phases/SF6/Draw
Atoms tab;
the list will be shown and the two unique atoms drawn on the plot.


To improve
this do the following:
1)
Double click the Style column heading and select ellipsoids; the figure will be redrawn
showing the 50% ellipsoid surfaces.
2)
Double click the empty upper left box in the Draw Atoms table; both atoms will turn
green and the two table rows will be grey.
3)
Under the menu Edit Figure select Fill unit cell; the figure will be redrawn
showing all atoms in the cell (too many bonds, though).
4)
Double click the Type column heading and select the S atom type
5)
Under the menu Edit Figure select Fill CN-sphere; the figure will be redrawn
with six F atoms about all S atoms (still too many bonds).
6)
Finally select Draw Options tab and change Bond search factor to 0.7; the figure will be redrawn
to show

which
is what one expects for orientational disorder for the SF6
molecules. Next we will explore this with a RMCProfile simulation. To keep this
drawing, you may want to save the GSAS-II project.
Reverse Monte Carlo
Simulation of SF6
RMCProfile
is most effective if a large box is used for the modelling; this requires very
long running times (10-20 hrs for SF6) before a meaningful result is
obtained. However, for the purposes of this tutorial, we will be using a
smaller big box model that converges in a more reasonable time (~10min). The
result will clearly fit the data but the model is too small to give enough
molecular orientations to be meaningful, however this exercise will show you
how to set up RMCProfile from within GSAS-II.
To
start select the Phases/SF6/RMC tab; if rmcprofile.exe is
within the GSASII directory the data window will show
At
the top is a radio button selection for RMCProfile and fullrmc. The latter is an
alternative big box modeling program (not working – under development);
RMCProfile is selected by default and all below are its setup controls. There
are four major sections (metadata, major controls, restraints & constraints
and data controls); we will work through each of these in turn. The information
you enter here is retained in the GSAS-II project so you can easily try
alternative setups without having to enter everything over again.
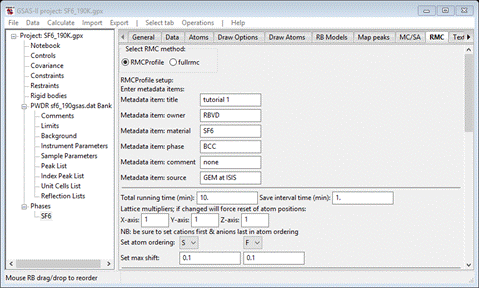
Step 1. Set metadata
items
The
entries here are for your convenience; there is no explicit use made of any of
these, but they will appear in some of the output files from RMCProfile and of
course will show here on subsequent views of this window. Fill out as many of
them as you see fit. I entered some things for each as the defaults are
somewhat nonsensical.
Step 2. Set general
controls
The
running time is defaulted to 10 minutes with a Save interval of 1 minute. At each save time a
number of files are written by RMCProfile; these can be viewed by using the
Operations/Plot command (more about this later – don’t bother trying it now,
there is nothing to see). For the purposes of this tutorial leave them at their
defaults.
The
big box model used by RMCProfile is described as multiples of the unit cell
axes. In this case we want a 3x3x3 box so enter 3 for each of X-axis, Y-axis and Z-axis.
Next
is to set the order of the atom types in the structure; this is used to
construct atom-atom distance restraints on the modelling. The order here (S followed by F) is appropriate; if changed
the window will repaint updating various entries possibly resetting some
entries to defaults. I suggest you decide the order now and then don’t change
it later. Set the maximum shift for S to be 0.05 (the value for F is OK).
When done the window should look like
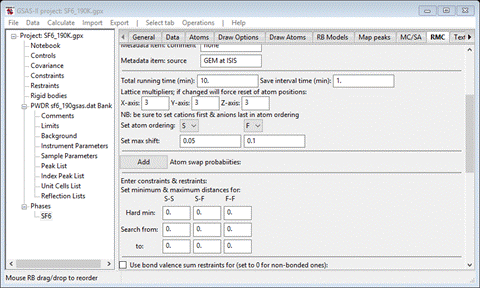
Skip
the next item (Atom swap probabilities). This is for cases in which atoms can
exchange sites.
Step 3. Set
constraints/restraints
Next
is to set the “Hard” minimum atom-atom separation and the allowed search rage
for each pair. Enter 4, 1.37 and 2 for the S-S, S-F and F-F Hard min. This rejects any proposed atom move that results in a
contact less than that value.
The
search range further restricts the allowed moves; this can maintain bonding
within the structure. Enter 1.37 & 1.74 for the S-F Search from and to values, and 2.0 & 2.42 for the F-F values. The window should
look like

Scroll
down to the bottom of the window for the last section.
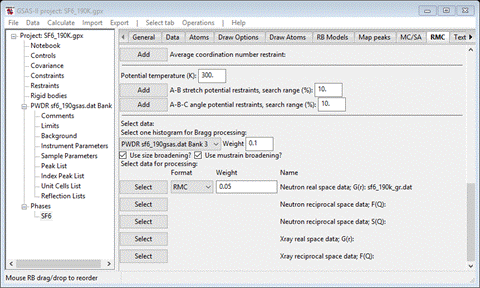
Step 4. Select data
to be fitted
We
will be using three different types of SF6 data for the fitting by
RMCProfile. The first is the selection of the powder pattern (“Bragg”) for
processing. This is taken from the PWDR entries in the GSAS-II project and
accessed from the pulldown; there is only one “PWDR sf6_190gsas.dat Bank 3” that was used earlier in
your Rietveld refinement of SF6. If you had used multiple banks in
the Rietveld refinement, all would be shown in the pulldown, but only one can
be selected. Set the weight to 0.1 (NB: smaller numbers means a heavier weight).
Next
press the Select button for the “Neutron real
space data; G(R)”
line; a FileDialog should appear. Because you used the Help/Download tutorial menu
entry to open this page and downloaded the exercise files (recommended), then
the RMCProfile-I/data/...
entry will bring you to the location where the files have been downloaded. (It
is also possible to download them manually from https://advancedphotonsource.github.io/GSAS-II-tutorials/RMCProfile-I/data/.
In this case you will need to navigate to the download location manually.)
The
FileDialog should show

Select
sf6_190k_gr.dat; it will be copied from this
location to your working directory for this tutorial (RMCProfile requires all
its files to be local). The window will be redrawn showing the new entry; the
weight is fine.
Next
press the Select button for “Neutron reciprocal space data: F(Q)”; the FileDialog will show
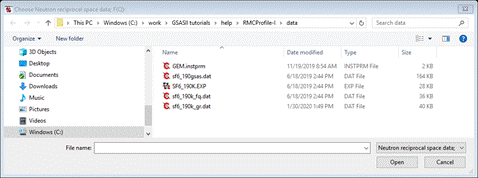
Select
sf6_190k_fq.dat; again it will be copied to
your working directory. The window will be redrawn
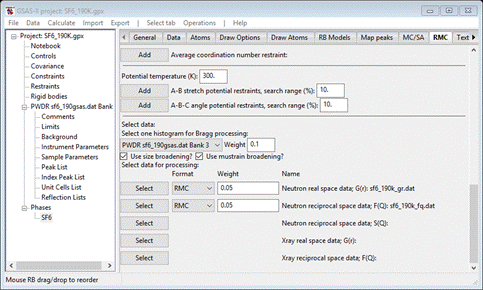
Change
the weight to 0.01. Your local directory should have just 3 files

Step 5. Setup RMCProfile
files
You
are now ready to setup the RMCProfile input files.. Do Operations/Setup
RMC from the
menu; the console should report that files were written
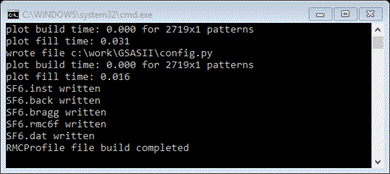
and
your local directory should now have 8 files
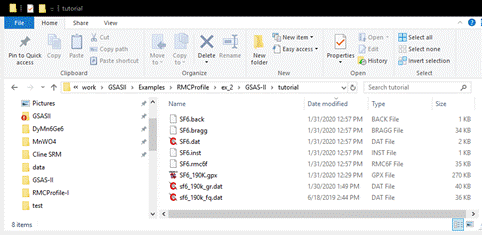
These
text files contain data needed by RMCProfile for the fitting of SF6. They are:
SF6.back- the 15 coefficients for the
chebyschev-1 function needed to comput the background for the Bragg pattern
SF6.bragg – the powder pattern used in
the Rietveld refinement
SF6.dat – the RMCProfile controls
file; described in full in the RMCProfile User Manual. It can be edited if need
be, but remember it is rewritten each time Operations/Setup RMC is done.
SF6.inst – the instrument parameter
coefficients for the neutron TOF peak shape function used by GSAS-II and
implemented in RMCProfile for computing the Bragg pattern.
SF6.rmc6f – the big box set of atom
positions. It is normally not rewritten when Operations/Setup RMC is done
unless the X-axis, Y-axis, or Z-axis lattice multipliers are changed. Most
important is that it will contain the set of big box atom positions as updated
by RMCProfile according to the Save interval (every 1 min in this case).
You
are now ready to run RMCProfile.
Step 6. Run
RMCProfile
To
run RMCProfile from inside GSAS-II, do Operations/Execute. You will first see a “nag”
note asking you to cite the publication describing RMCProfile; press OK.
The
program will start in a new console window – processing will initially be
pretty fast for this case and then slow down as the modelling proceeds. It
reports Time used and Last saved. Once the latter is nonzero you can view
intermediate results to see its progress. Note that you can exit GSAS-II and
RMCProfile will continue running. After RMCProfile finishes note that the
project directory now has ~30 files many of which are just temporary ones
created by RMCProfile. We will be looking at just the *.csv files and the
SF6.rmcf6 file; the latter contains the last atom configuration acceptable by
RMCProfile thus representing a best disordered model.
Step 7. Viewing
results from RMCProfile
Do Operations/Plot; a FileDialog showing only
*.csv files will appear
They
all should have the same prefix in their name, “SF6” which is the phase name
from the GSAS-II project file. Select any one of them – it doesn’t matter. All
of them will be read and their contents displayed as individual plots. For
example the chi^2 plot shows the progress of the RMCProfile fit

The
inset is the upper 3/4ths of the plot magnified; you can see that RMCProfile is
still improving the fit slowly even after 10 minutes of running.
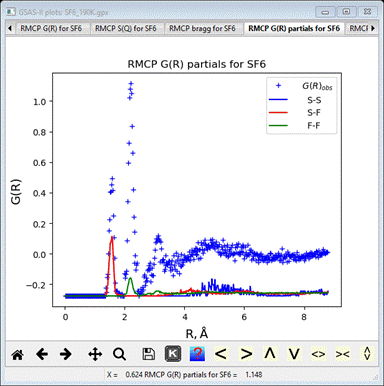
The G(R)
partials plot shows the identity of each feature; the first 2 sharp peaks arise
from S-F (1.58Å) and F-F (2.20Å) distances within the SF6 octahedron
(cf. 1.457Å and 2.06Å, respectively, for the average structure) while the
broader one at 2.8-3.1 are from F-F intermolecular contacts.
Comparing the Bragg plot with the PWDR plot shows that the
RMCProfile fit is very similar to the Rietveld fit
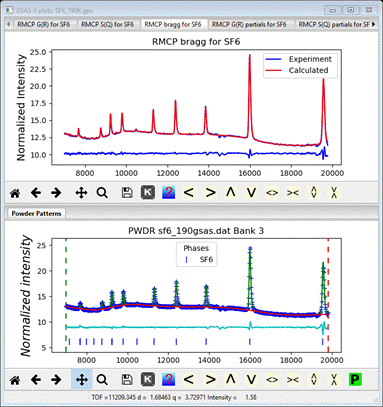
This
plot was made by dragging one plot tab to the bottom of the screen to create
the second frame.
Step 8. View the
big box structural model for SF6
Next,
we can view the resulting big box structure. Do Import/Phase/from RMCProfile
.rmc6f file;
a FileDialog box with one entry will show the required file, SF6.rmc6f. You will first see a Is this the
file you want
popup window; select Yes. Next will be an Edit phase name popup. The proposed name is
the same as the existing one; GSAS-II will rename this one by adding ‘_1’ to
the end. Next will be a popup for Add histograms; respond Cancel because you don’t want this
phase to be in any subsequent Rietveld refinement. Looking at the General tab
for this phase we see it is 3X in all three axes relative to the original and
has no symmetry (space group P 1).
Select
the Draw
Atoms tab
for this phase; a van der Waals ball model will be drawn. You can select the S atoms & fill the CN-sphere for them (does take time –
be patient) and then change all the atoms to ball and stick style. The
CN-sphere filling works because the RMCProfile structure still has
translational symmetry across its 3x3x3 lattice. You should see something like
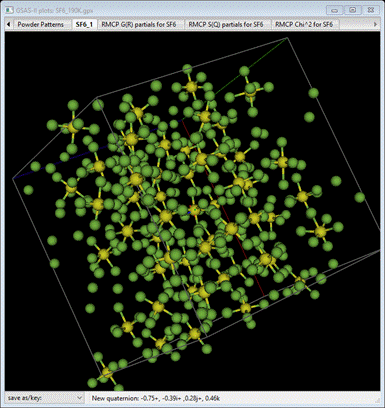
Notice
the rotational disorder as well as some positional shifting of the SF6
molecules. The stray F atoms are bound to SF6 molecules in
neighboring boxes.
NB:
this import facility can be used to load any rmc6f file, e.g. from a RMCProfile
run done outside of GSAS-II; all the plotting and tools in this and the
following steps are available for these big box models.
Step 9. View the
disordered average structure
We
can compress the big box result back into the original average crystal
structure to see how the disordered sites compare with the average ones. Select
the General tab for the big box phase
(SF6_1) and then do Compute/Transform. A popup dialog will appear
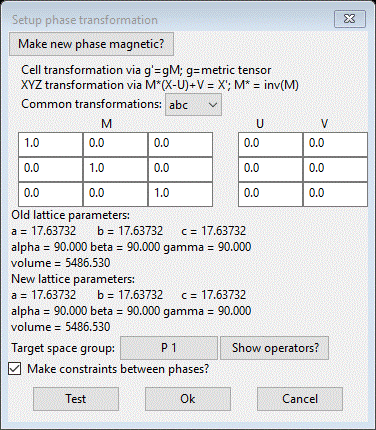
This
is the general tool inside GSAS-II for all sorts of structure transformations.
Here we will use it to push all the big box atoms back into the average
structure unit cell. Recalling that the big box model is 3x3x3 the original
unit cell, we simply want to reverse the process. Enter 1/3 into the diagonal elements
of the M matrix. The GUI will convert them to the decimal equivalent (0.333…).
The target space group should be P 1. You should see
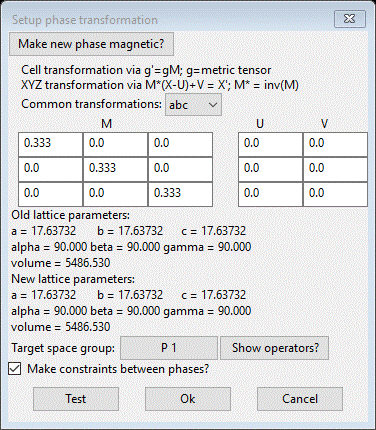
Press
Ok to do the transformation. A
new phase, “SF6_1 abc” will be formed as triclinic with cell axes that match
the original cubic ones. Select Draw Atoms; a van der Waals model will appear.
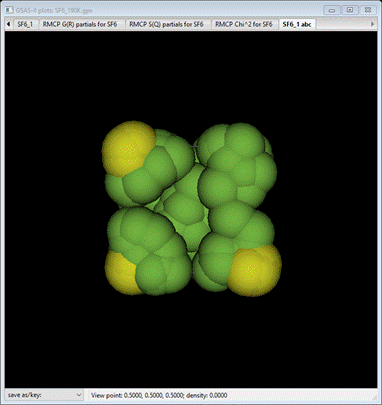
Select
the Draw
Options tab
and reduce the van der Waals scale to about 0.05.

This
compares pretty well with the original structure with the ellipsoids drawn at
90% probability.

Step 10. Polyhedral
comparison for the big box model
In
this step we will examine how the suite of SF6 molecules deviate
from an ideal octahedron. This analysis is currently restricted to structures
with P1 space group symmetry; this is what we have from Step 8 above. Select Phases/SF6_1 from the data tree; the General
tab will appear.

What
we want is at the bottom of this panel; scroll down to the bottom
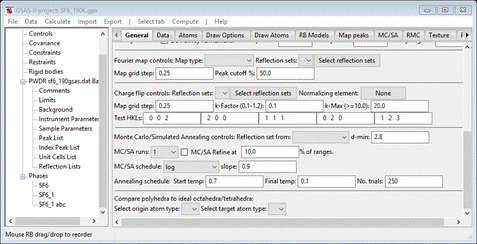
At
the very bottom is the control for polyhedral comparison. All that is needed is
to select the central atom (“origin atom type”) and the polyhedral
vertices (“target atom type”). Select S for the former and F for the latter. Then do Compute/Compare from the menu; a popup for
setting Distance Angle Controls will appear
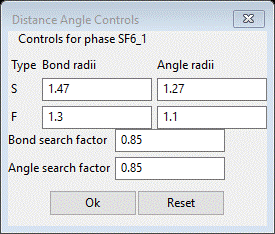
In
some cases the bond search factor may need to be adjusted to get the right
number of vertices for the polyhedral; in this case 0.85 is fine (NB: in this use the
angle ranges are ignored). Press OK; a progress bar will show the atoms being processed. The
console may show some atoms being skipped because the number of vertices wasn’t
either 4 (tetrahedron) or 6 (octahedron); all are ok here. In this case it
quickly finishes; for larger big box models this calculation can take a number
of minutes. The General tab reappears already scrolled to the bottom
A
new button (Show Plots?) has appeared after the atom selections; press it. A number
of new entries will appear on the plot window; we will discuss each in turn.
Select the Bond tab
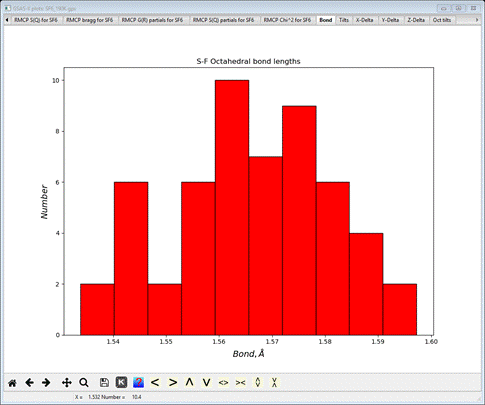
This
gives the distribution of S-F bond lengths for all the polyhedral found across the
big box model. It is rough because of the small size of the model. The console
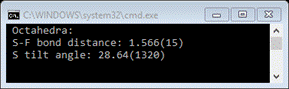
will have listed the average value with the standard uncertainty in
parentheses.
Next
select the Tilts tab
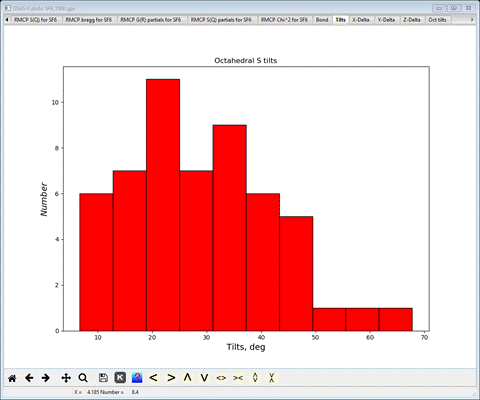
This gives the distribution of axial tilts of the SF6
molecules from the reference octahedron (aligned along the Cartesian axes).
There is a wide distribution with a roughly average tilt of ~30 deg (the
average tilt is also shown on the console). Next select X-Delta, Y-Delta and Z-Delta tabs in turn.
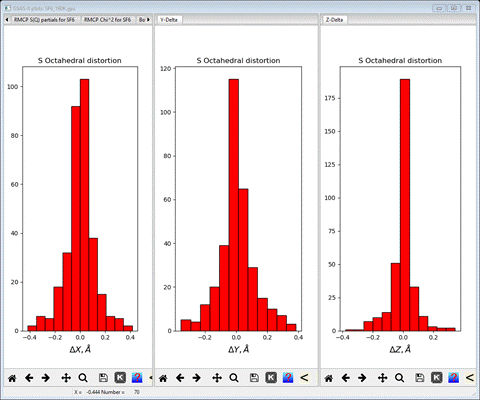
I
have made this plot by dragging the respective plot tabs to one of the right
(or left) edges to create a 3 pane plot. These show the displacement of the F
atoms from the ideal octahedral vertices along each Cartesian axis. Here they
are arranged about the zeros fairly tightly. If there were structural distortions
to the octahedra (e.g. Jahn-Teller distortions) these could show in these
plots. Finally select the Oct tilts tab; a 3D plot will be shown

The
sticks marks show the directions of the unit vectors about which the SF6
molecules are tilted with respect to the reference octahedron and the colors
indicate the angle of rotation according to the color bar on the right. In this
case they are quite random; they cover all directions and rotations. Note that
the algorithm selects the shortest F atom to octahedral vertex for the tilt
calculation. That could be any one of the six possible along +/- xyz axes.
This
completes this RMCProfile tutorial; you should save the project.
Final note
A
production run with enough atoms to give decent statistics would be for a box
that is 10x10x10 the original unit cell and would require a 10-20 hour run
time. It would contain ~14000 atoms instead of 377 as used here.