PDFfit-III
Introduction
In
these tutorials, we show how to use GSAS-II in conjunction with PDFfit2 (“PDFfit2
and PDFgui: computer programs for studying nanostructures in crystals",
C.L. Farrow, P.Juhas, J.W. Liu, D. Bryndin, E.S. Bozin, J. Bloch, Th. Proffen
& S.J.L. Billinge, J. Phys, Condens. Matter 19, 335219 (2007), Jour. Phys.:
Cond. Matter (2007), 19, 335218. doi:
https://doi.org/10.1088/0953-8984/19/33/335219) to fit pair distribution
functions (PDF) with a “small box” disordered model. We will refer to PDFfit2
as “PDFfit” in these tutorials as well as in the GSAS-II user interface for it.
The
GSAS-II interface to PDFfit is a simplification where some capabilities of
PDFfit are not implemented; GSAS-II only allows a single phase for PDFfit and
the atom thermal displacement model is strictly isotropic. PDFfit allows
multiple phases and anisotropic thermal motion; we considered these to be not
useful and an unneeded complication, and these are perhaps better tackled via
Rietveld refinement with the original diffraction data.
We
have included with the GSAS-II distribution the most recent version of the
PDFfit2 executable and its python interface routines for Windows (those for Mac
OSX and linux will follow in due course). They are in the diffpy subdirectory of GSAS-II. No
other files are needed. Also, the diffpy/manual subdirectory has the paper
referenced above as well as one that described the original Fortran version of
PDFfit. PDFgui.html describes 3 tutorials, upon
which the GSAS-II ones are based, as given for the PDFgui software.
Alternatively, you can install pdffit2 from Anaconda by executing
conda install -c diffpy diffpy.pdffit2
in a console
window after activating your version of python.
It will be installed in your python as
gsas2full/Lib/site-packages/diffpy.
In
this tutorial we will continue the analysis LaMnO3 using variable
temperature neutron G(r) patterns to introduce you to the process of doing
sequential PDFfit analysis of mode distortions within GSAS-II. At the end you
will be able to step through the sequence and see how the structure responds to
temperature change from 300K to 980K. If you haven’t done so already, start
GSAS-II.
If
you have already done the PDFfit-II tutorial and have retained the result from
ISODISTORT (LaMnO3-Pbnm_child.cif), you can skip Steps 1 &
2 and go directly to Step 3, because these duplicate your operations in that
tutorial to yield this cif file.
Step 1. Setup LaMnO3
Pbnm phase
The
blank startup view of GSAS-II begins with this
First,
we need a description of the expected LaMnO3 structure (probably
generated from a powder or single crystal structure determination); this is in
the data package for this tutorial. Do Import/Phase/from
CIF file.
A file dialog box will appear. Navigate to PDFFIT-III/data and select LaMnO3 Pbnm.cif. Respond Yes to the file check box and accept the phase name as given.
The General tab will appear
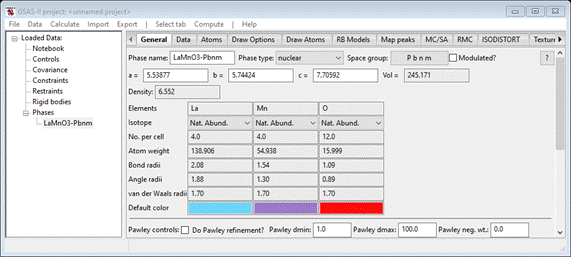
If
desired, you can examine the 4 atom positions and make a drawing by following
the operations as outlined in the previous tutorial.
Step 2. ISODISTORT
analysis of LaMnO3
If
you access the ISODISTORT web site directly via https://iso.byu.edu/isodistort.php,
it will take you through a sequence of web pages with many possible
alternatives to choose from, but it always begins with an undistorted high
symmetry parent structure. We have reduced all this to a single preselected
step that compares a given structure (e.g. this LaMnO3 Pbnm phase)
and compares it to the undistorted parent phase and determines the possible
distortion modes and their magnitudes (ISODISTORT Method 4). This operation
results in a cif file containing all the information required by GSAS-II to
construct a mode refinement model. To begin select the ISODISTORT tab for the LaMnO3-Pbnm phase; you should see
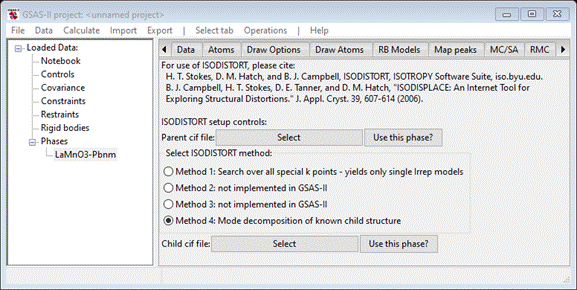
Press
the “Parent cif file” Select button; a file selection
dialog will appear. Navigate to PDFFIT-III/data and select LaMnO3 Pm3m.cif. The window will repaint to
show the choice (note that this does not import this phase into GSAS-II). Since
we have already imported the Pbnm phase of LaMnO3 and are working
out of its ISODISTORT tab, we can use it for the “Child cif file”. Press
Use this phase? The window will repaint
giving the two choices.
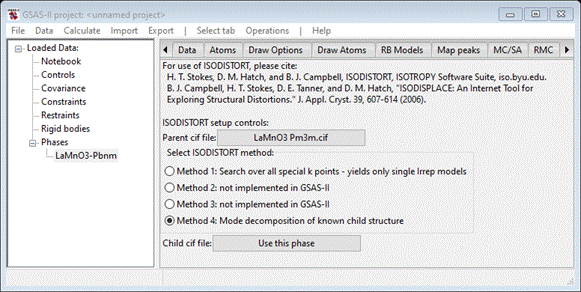
We
are now all set to run ISODISTORT; you must be connected to the internet for
this to work as we are remotely accessing the https://iso.byu.edu/isodistort.php
web page and all that follows (about four more pages). Do Operations/Run ISODISTORT; the console will show a series
of messages as GSAS-II accesses various web pages at the ISODISTORT site. It
will successfully finish with a popup message
Press
OK; the console will display a
final message
indicating
that a new cif file (LaMnO3-Pbnm_child.cif) has been created. This contains all
the constraint information needed to construct them in GSAS-II.
Step 3. Import and
examination of ISODISTORT result
If
you still have LaMnO3-Pbnm_child.cif from the PDFfit-II tutorial,
you can start here. Do Import/Phase/from
cif file;
a file dialog box will appear. There will be (at least) 2 files. One is the one
created for input to ISODISTORT (ISOin.cif) and the other is the result from
ISODISTORT, LaMnO3-Pbnm_child.cif. Select it; respond Yes to the popup file check and
leave the phase name as it is. The console will display several messages as
GSAS-II interprets the content of the cif file and develops the appropriate
constraints to make the distortion modes as GSAS-II variables. These are then
displayed in the ISODISTORT tab
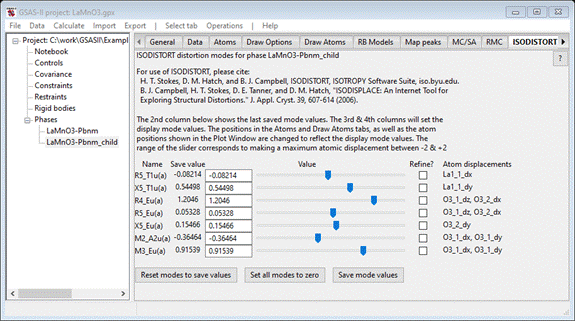
Shown
are the 7 displacement modes (NB: the same number as the number of independent
atom coordinates in this Pbnm perovskite), their values in Angstroms and at the
end the atom coordinates involved in each one. If you began with the LaMnO3-Pbnm_child.cif file from PDFfit-II tutorial skipping Steps 1
& 2, there will only be one phase in the project.
To
see how the various modes change the structure, you must draw it first; this
will be useful to have when you examine the structure to see how it changes
with temperature. (Trying the sliders now will give you an error message).
Select the Draw Atoms tab
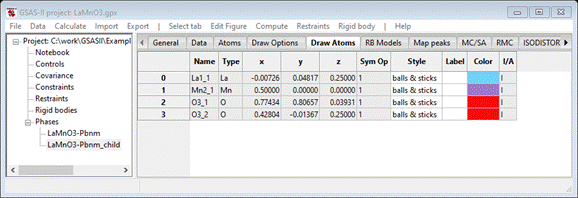
Select
all atoms, do Edit
Figure/Fill unit cell.
Then left double click Type and select Mn; then do Edit
Figure/Fill CN-sphere.
You should see (after a bit of rotation)
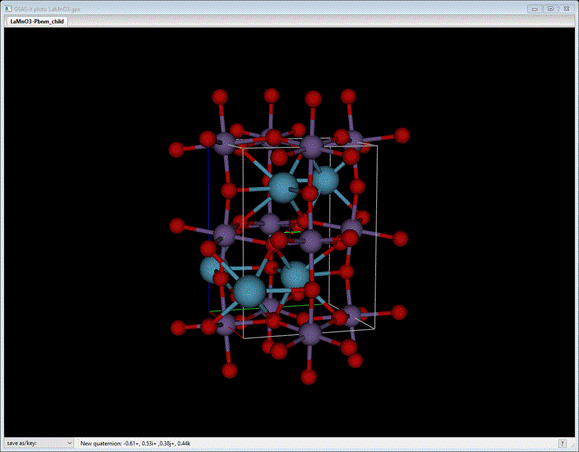
Now
select the ISODISTORT tab. You can now shift the sliders
to see what each mode does to the structure. When done, press the Reset modes to save values to recover the original
structure. The tab should look like
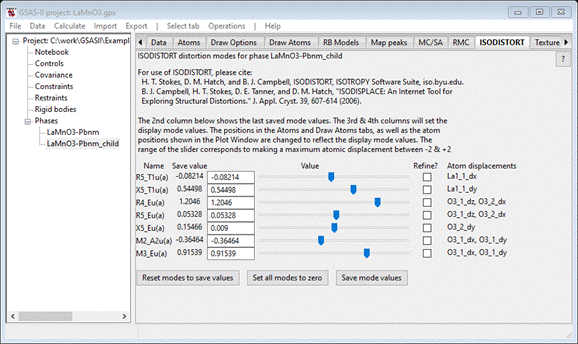
This
would be a good time to save the project (do File/Save project); use a name that is
different from the one used in the PDFfit-II tutorial (I used LaMnO3 seq.gpx).
Step 4. Setup for
PDFfit
We
now need to convert this phase (LaMnO3-Pbnm_child) into one that is setup for
PDFfit. This process creates a new phase with the constraints transformed into
PDFfit constraints. From the ISODISTORT tab, Do Operations/Make PDFfit phase; the window repaints with
the ISODISTORT tab still selected
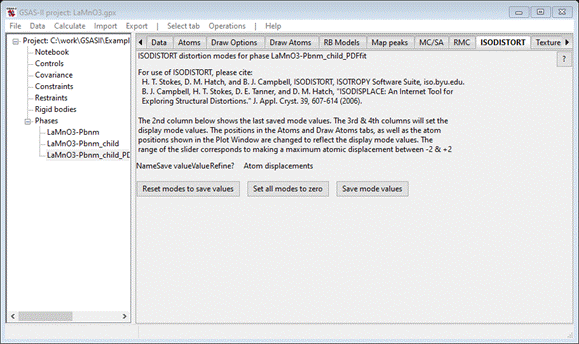
This
is now the third phase (LaMnO3-Pbnm_child_PDFfit); the mode information
has been transferred. Select the RMC tab and choose the PDFfit radio button; the window now displays the full suite of
atom PDFfit position constraints as developed from the ISODISTORT displacement
modes.
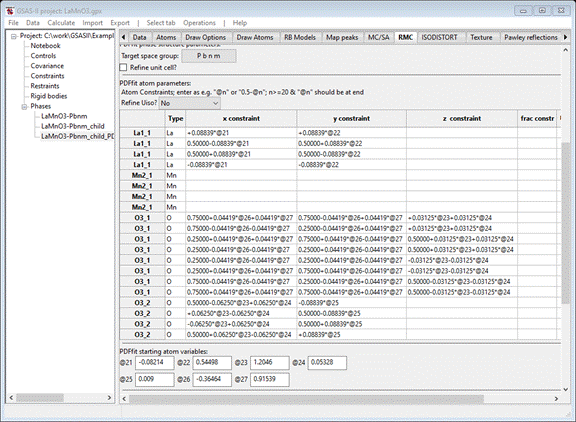
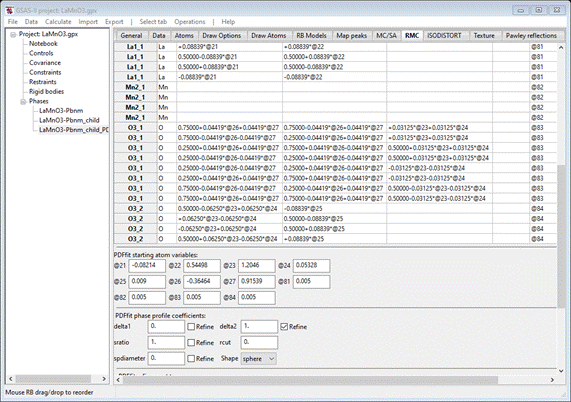
The
atom variables (“@21”, etc.) correspond directly with the ISODISTORT distortion
modes shown for the LaMnO3-Pbnm_child phase; their values are shown as well.
The correct target space group is given (P b n m); you should check the Refine unit cell? box. You should also choose a Uiso refinement
model. I selected Refine Uiso ?
by parent name,
that gives 4 additional parameters (“@81” – “@84”) with starting values
(Uiso=0.005) as listed. As with the two earlier tutorials, change “delta2” to 1.0 and check its Refine box. When done the RMC tab
should look like
Because
the only phase needed for the rest of this tutorial you can delete the others;
do Data/Delete phase entries from the main menu. A popup
will appear
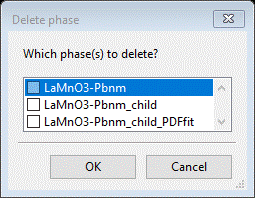
Select
the 1st two items & press OK. The window will be
repainted showing only one phase in the data tree
Save
the project. (I had used LaMnO3
seq.gpx)
Step 5. Reading
sequential PDF data for LaMnO3 into GSAS-II and sequential PDFfit
setup
The sequential
PDFfit operation requires that the G(r) data be given as a suite of “PDF”
entries in the GSAS-II tree. Do Import/PDF
G(R) Data/from r(A) step G(r) data file; a file selection dialog will appear. Navigate to
PDFfit-III/data and select all 11 gr files; the window should
show all 11 entered in the tree. (Notice that I have deleted the other two
phases).
The
plot may show G(r) for the last one read.
Now
select the LaMnO3-Pbnm_child_PDFfit phase; it should show the
RMC tab with PDFfit selected. The bottom of this window should show
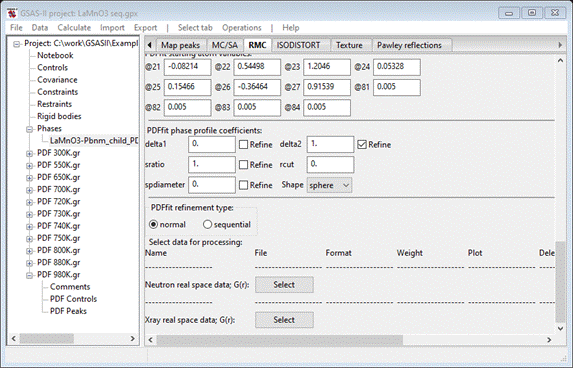
Select
sequential for the PDFfit refinement type;
the window will be redrawn. The bottom now shows
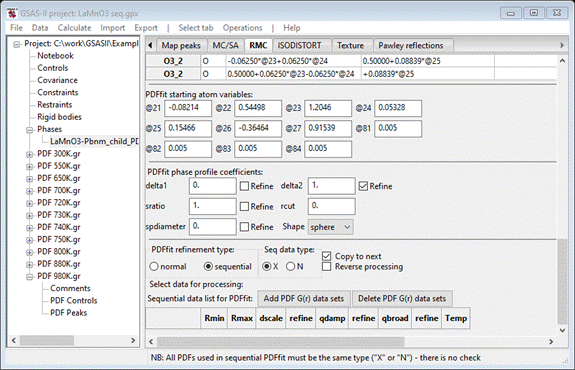
Change
Seq data type to ‘N’ and then press the Add PDF G(r) data sets button; a popup dialog will
appear
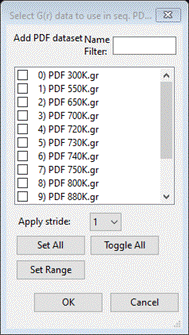
Press
Set All and OK; the window will be redrawn showing a table of the data
sets at the bottom
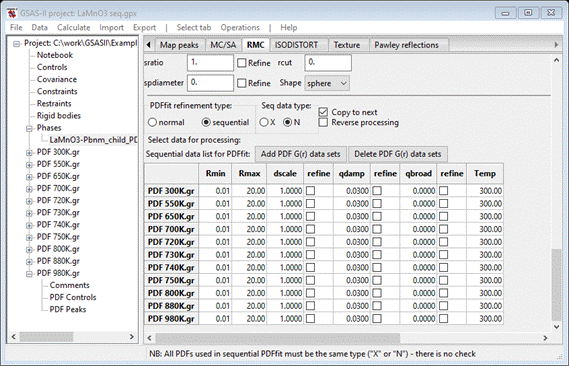
Unfortunately,
the data sets did not (apart from the file name) contain a temperature record.
You’ll have to enter them by hand. Notice the fine stepping in temperature
around 700-750K; this was to track some special structural change in this
material. We will be keen to see what comes out of the mode displacements.
We
need to set Rmin to 2.0; double click on the Rmin column heading. A small popup will appear; set the value to
2.0 & press OK. The window will be redrawn. Now do the same with the refine column headings for dscale and qdamp selecting Y – vary all in each case. The window is
refreshed each time (you’ll have to scroll back down to the bottom each time).
When done, this part of the window should look like
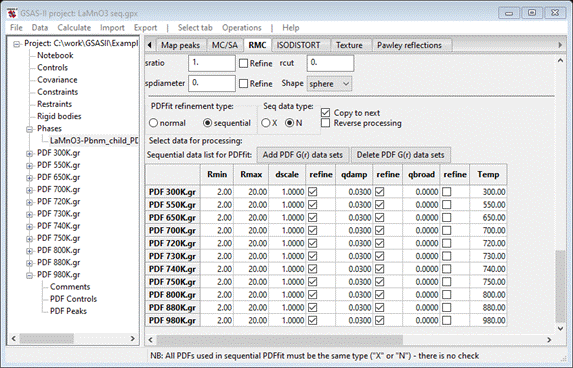
Note
the Copy to next box is checked; this
facilitates the initial sequential refinement. This finishes the initial setup
for a sequential fit using PDFfit. You should save the project.
Step 6. Sequential distortion
mode PDFfit for LaMnO3 with temperature
To
begin, we must setup the for the sequential PDFfit run. Do Operations/Setup RMC; a short message will appear
on the console indicating that Sequential_PDFfit.py written; this is the python script that
will drive PDFfit.
Next,
do Operations/Execute; a popup will appear
reminding you to properly cite your use of PDFfit (don’t forget to cite GSAS-II
as well). Press OK. A sequence of consoles will
appear, one for each execution of PDFfit. A progress bar will show
Most
steps will be quick (a few seem to require more cycles to converge), but all
will show Rw ~ 10%. When finished, the progress bar will vanish, and the
sequential result is shown
Step 7. Examination
of sequential PDFfit result
There
are many ways to examine a sequential result in GSAS-II. Simplest is to select
a column heading; the plot will show the parameter as a function of run number
(I picked 11-a – the unit cell axis a).
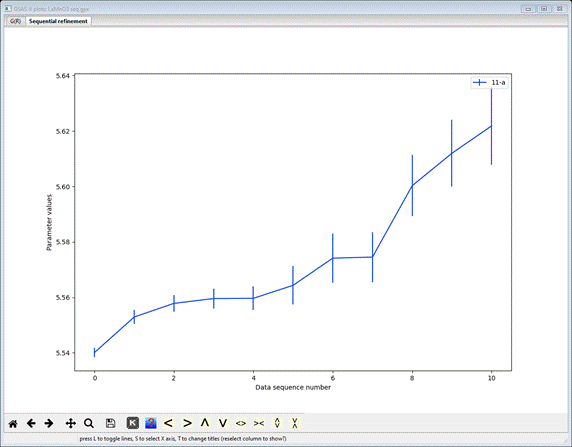
The
vertical bars represent the standard uncertainties for each a-axis
determination by the sequential PDFfit. Select the plot window and press the ‘s’ key; a small popup window will appear
Select
Temperature and press OK; the plot will be redrawn with sample temperature as the
x-axis
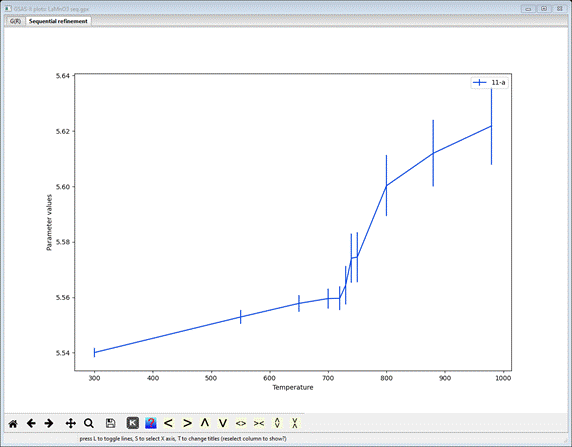
This
is considerably more informative. Something interesting seems to be happening
at ~750K. Now select all the mode distortion parameters – pick 21-R5_T1u(a) and then hold the Ctrl key down while you pick the rest (to 27-M3_Eu(a)); the plot will show
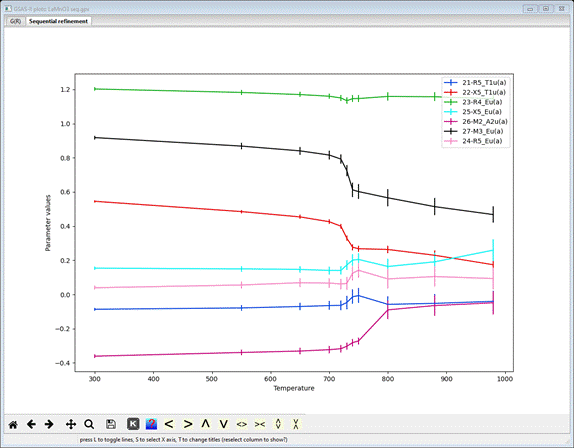
Clearly
the anomaly on the a-axis lattice parameter is coincident with distinct transitions
in most of the mode distortions. The other thing to note is the trend toward
zero for all but the R4_Eu(a) (green curve) mode; it remains essentially
constant over the entire temperature range. The convergence of some of the
modes prestages the orthorhombic to rhombohedral phase transition at ~1000K for
which there are fewer possible distortion modes.
To
see the effect of these modes on the structure, GSAS-II allows one to step
through the sequence while viewing the crystal structure. Select the Draw Atoms tab for this phase. Then
fill it out using the Fill unit
cell and
Fill CN-sphere commands. The initial view
may be
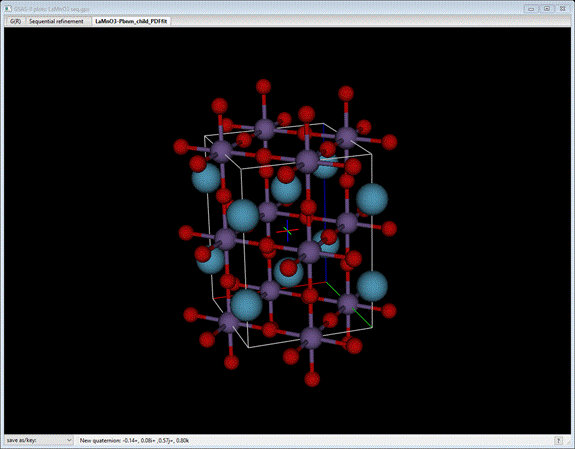
which
is the undistorted parent LaMnO3 cubic perovskite transformed to the
orthorhombic Pbnm lattice. Now press the +/= key; the drawing will change
to show the effect of the distortion modes at 550K (see message at bottom of
plot)
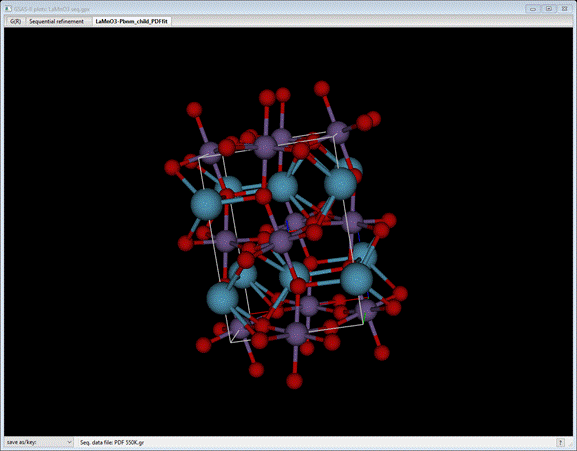
Continue
to press the +/= key (or the _/- key; it just steps in the opposite direction) while viewing
the structure from various vantage points. The message will tell you what the
current set of parameters is. You can recover the parent structure by doing Edit atoms/Update draw atoms from the Atoms tab.
When
finished, you may save the project. This concludes the sequential PDFfit
tutorial.