PDFfit-I
Introduction to use of PDFfit in GSAS-II
Introduction
In
this and subsequent tutorials, we will show how to use GSAS-II in conjunction with
PDFfit2 (“PDFfit2 and PDFgui: computer programs for studying nanostructures in
crystals", C.L. Farrow, P.Juhas, J.W. Liu, D.
Bryndin, E.S. Bozin, J. Bloch, Th. Proffen & S.J.L. Billinge, J. Phys,
Condens. Matter 19, 335219 (2007), Jour. Phys.: Cond. Matter (2007), 19,
335218. doi: https://doi.org/10.1088/0953-8984/19/33/335219) to fit pair
distribution functions (PDF) with a “small box” disordered model. We will refer
to PDFfit2 as “PDFfit” in this & subsequent tutorials as well as in the
GSAS-II user interface for it.
The
GSAS-II interface to PDFfit is a simplification where some capabilities of
PDFfit are not implemented; GSAS-II only allows a single phase for PDFfit and the atom thermal displacement model is strictly
isotropic. PDFfit allows multiple phases and anisotropic thermal motion; we
considered these to be not useful and an unneeded complication, and these are
better tackled via Rietveld refinement with the original diffraction data.
Note that PDFfit2 is not installed with GSAS-II by default, but with usual installation configurations, where GSAS-II is run from a conda-installed version of Python, it can be installed for you (see Step 3, below).
In
this tutorial we will use a simple analysis for Ni using neutron G(r) and x-ray
G(r) patterns to introduce you to the process of doing PDFfit analysis within
GSAS-II. If you haven’t done so already, start GSAS-II.
Step 1. Setup
parent Ni phase
The
blank startup view of GSAS-II begins with this
Do Data/Add
new phase
and enter Ni to replace “New phase” in the popup. The window will be redrawn
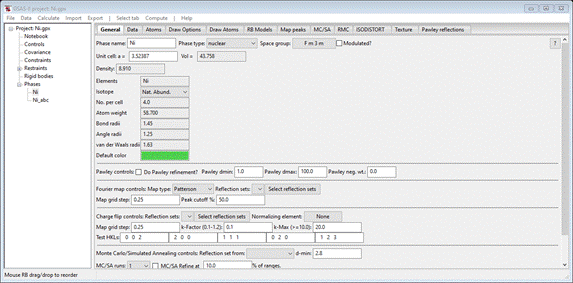
Change
the space group to F m 3 m (don’t forget the spaces). This will be done in a popup and
a second popup will show the operators for Fm3m; the window will be redrawn to
show the required entry for the unit cell. Enter 3.524 for Unit cell a; the window
should show
Now
select the Atoms tab; it will be empty
Do Edit
Atoms/Append atom;
a dummy atom will appear
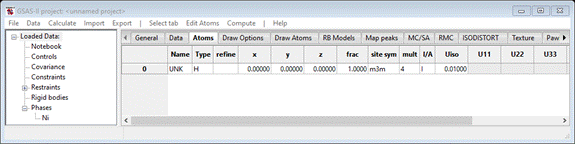
Double
left-click on the atom type (“H”), after a short pause a popup with a periodic
table will appear. Select Ni & press OK; the Atoms tab will be redrawn
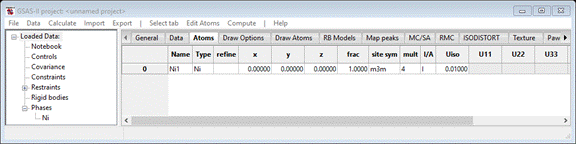
The
nickel atom is already in the correct position (0,0,0) so we are done setting
up the parent Ni phase.
Step 2. Make the
PDFfit phase
Because
PDFfit utilizes no symmetry information from the space group that is associated
with the structure to be studied, it requires a full unit cell of atom
positions. This step uses the Transform tool in GSAS-II to generate these as a
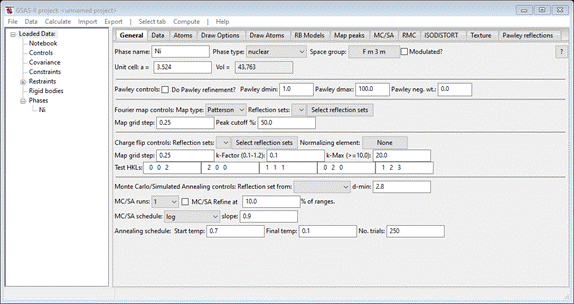
new phase. Return to the General tab for the Ni phase you should see
Do Compute/Transform; a popup will appear
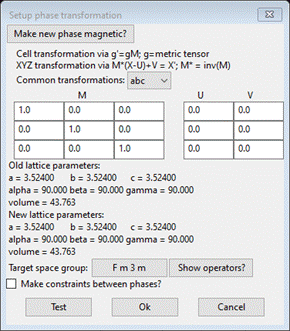
Change
the Target space group to P 1 and press Ok. A new phase with the name “Ni abc” will appear; this will
be used for the PDFfit analysis. The General tab for the new phase shows
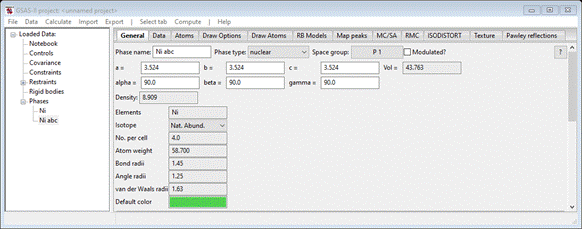
The
phase name “Ni abc” is ok for the rest of GSAS-II, but unfortunately must be
changed for PDFfit – it cannot contain any spaces, because it is used to create
various file names and PDFfit is not able to handle file names with spaces.
Change the phase name to “Ni_abc” (you can use anything else just so long as the name has no
spaces & isn’t “Ni”). The Atoms tab shows 4 atoms as the unit cell contents.
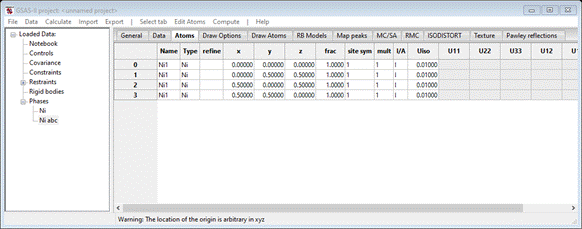
which
is the correct full cell description for nickel suitable for use in PDFfit.
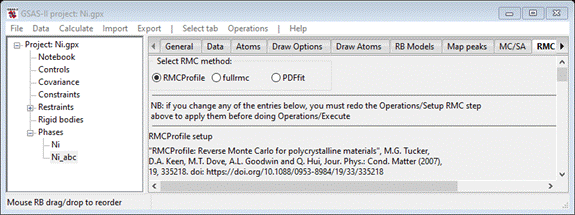
Step 3. Setup
PDFfit
All operations
for PDFfit are done in the RMC tab; select it and you should see the interface
for the default (RMCProfile)
Select the radio button at the top for PDFfit. If GSAS-II is running under a conda-installed Python, and PDFfit2 is not installed, you will be asked if you want to install it. (If you have installed Python yourself, you will need to install PDFfit2 either as a separate Python interpreter -- recommended -- or inside the Python used for GSAS-II. If the PDFfit2 package is installed into the same Python interpreter used by GSAS-II using conda or pip this invites potential conflict where PDFfit2 and GSAS-II require different versions of packages. At present this does not appear to be a problem, but this could arise in the future. The pdffit2_exec config variable described below is not needed when PDFfit2 is installed in the same Python used by GSAS-II. See links on the DiffPy package and/or PyPi for PDFfit2 for more information on installing PDFfit2 manually.
If PDFfit2 is installed in a separate Python installation, the
location of this is saved in GSAS-II configuration variable
pdffit2_exec. This is done for you if GSAS-II installs
PDFfit2.
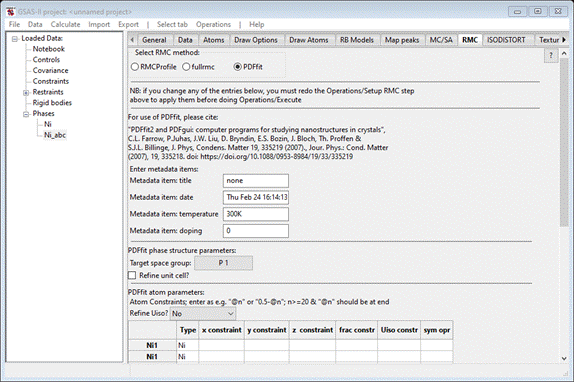
This
is the top half of the PDFfit interface and one sees the setup is based on the
Ni_abc phase (note the underscore you inserted in the phase name – this is
essential for correct PDFfit operation). You may enter items in the metadata
for your convenience; they play no role in the fitting by PDFfit. However, you
must change the Target space group from P 1 to F m
3 m so that proper symmetry is used for lattice parameter refinement
by PDFfit. Check the Refine unit cell box. Shifting to the lower
part of the interface we see
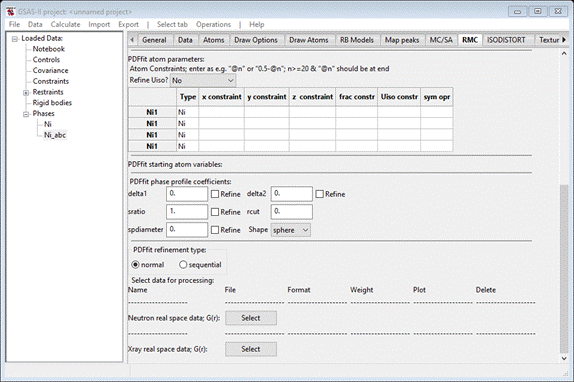
You
want to refine the Ni atom thermal motion parameters as a single value; select
“by type” in the Refine Uiso?
pulldown box. The window will be repainted showing “@81” for each of the Uiso
constr items in the table. This symbolism is used for PDFfit variables; the
code is “@N” where N is an integer. In GSAS-II we have assigned classes of
variables their own set of integers; we will see these after we run PDFfit fo
the first time.
The
coefficients “delt1” and “delt2” are for PDF sharpening effects due to
atom-atom correlated motion. They depend on 1/rij and 1/rij2,
respectively. Similarly, “sratio” is a PDF sharpening for correlated motion of
bonded atoms out to the distance limit “rcut” (not refinable). It is not likely that both descriptions will
be used simultaneously (might lead to singularities in PDFfit!). The remaining
term, “spdiameter” (Å), is a diameter for a nanoparticle sphere and is used for
damping the calculated PDF at larger R. If spdiameter = 0, this damping is not
used. Here we will be refining only delta2, set it to 1.0 & check it’s refine box.
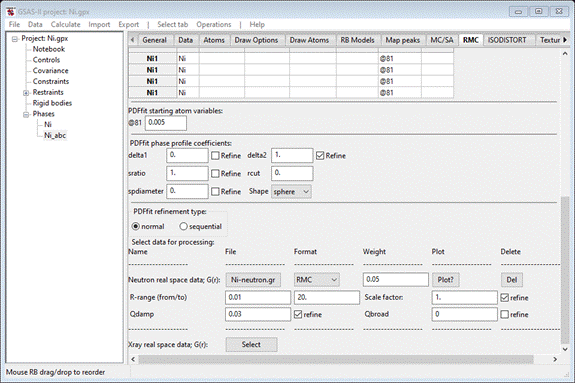
Next,
press the “Neutron real space G(r)” Select button; you will first be
asked to provide a GSAS-II project name (I used “Ni”) in a file dialog box. Next it will show another file
dialog box. Navigate to PDFFIT-I/data and select “Ni-neutron.gr”. It opens this file and
refreshes the RMC tab to show the new data set
If
you press the Plot? button, a plot of
Ni-neutron.gr will be shown
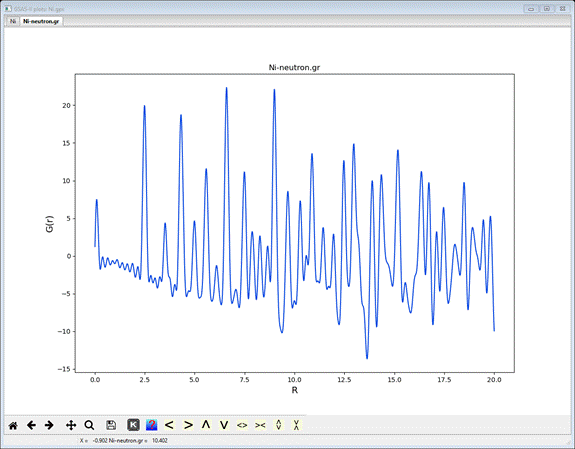
Note
that the 1st peak is at 2.45A; this is the nearest neighbor Ni-Ni
distance. You will want to set the value in the R-range 1st box to
be a bit below that (I used 2.0). (NB: PDFfit will crash if
this value is zero, so choose something suitable). The two PDF Gaussian broadening
coefficients: “Qdamp” depends on the limiting Q range of the original
diffraction data, and “Qbroad” depends on the noise level in the high Q
diffraction data. If zero, they are not used. We should use Qdamp=0.03 as a
starting value & refine it. The “scale factor” is nominally unity for PDF
data but can differ due to e.g. absorption effects in
the original data; thus it should be refined. When done the window should look like
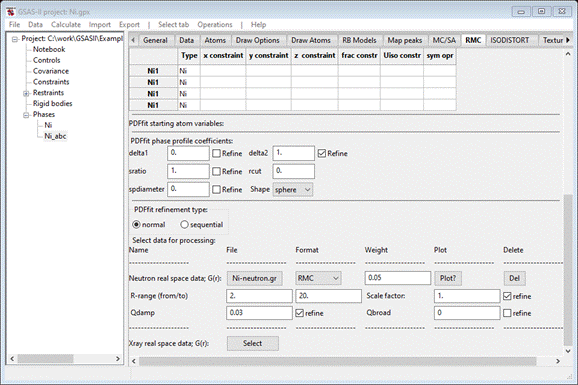
This
completes the initial setup for PDFfit. Save the project file.
Step 4. Running
PDFfit
PDFfit
executes by running in a separate process a short python script that contains
parameter setup values, refinement codes, etc. This is prepared automatically by
GSAS-II; do Operations/Setup
RMC from
the menu bar. A message will appear on the console and 2 new files will appear
in your working directory. They will be named “Ni_abc-PDFfit” with different
extensions (.py and .stru).
To
run PDFfit, do Operations/Execute from the menu bar. You will
first see a reminder to properly acknowledge your use of PDFfit (don’t forget
to cite GSAS-II as well!).
Press
OK. A new console window will
appear and run very quickly. A banner for PDFfit appears followed by setup
information and then its progress on refining the Ni structure. The end of the
display has convergence information (Rw = 6.975%; a good fit) and a
“Press any key to continue . . .” message.

Press
any key, the console will vanish, and a new popup appears
Press
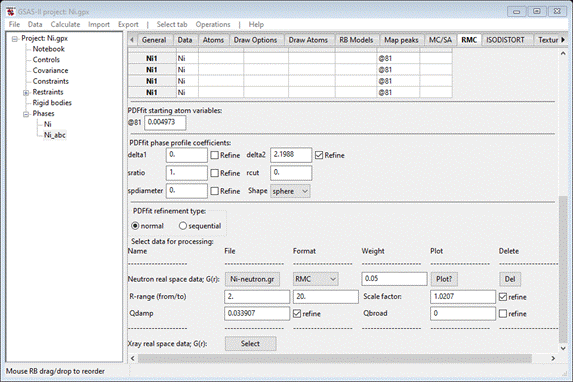
Yes, The RMC window is repainted
with updated values and some additional information (in the lower part)
If you do Operations/Plot from the menu, a plot
showing the fit will appear
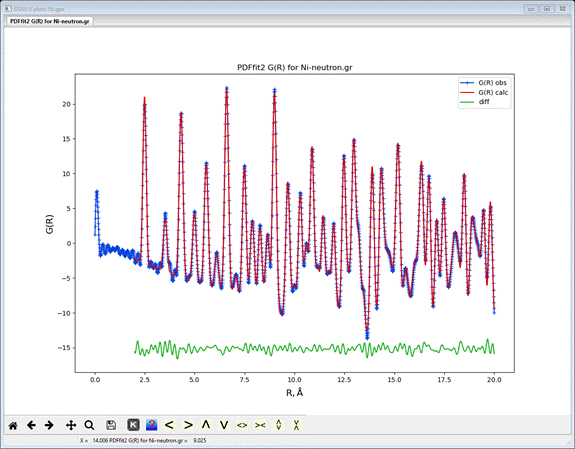
The blue curve is the original G(r) data, the red line is
the fit and the green line is the difference. Your working directory will have
3 additional files all named “Ni_abc-PDFfit” with the extensions “res”, “rstr”
and “fgr” (and an “N” in the latter file name). The first is a listing of the
setup information and results from the last refinement cycle.
Ni_abc-PDFfit.rstr is an updated version of the .stru
fine and contains updated phase and atom results with esds. The .fgr file
contains the observed and calculated PDF. Note that the lattice parameters in
the General tab have been updated and the Uiso values for the Ni atoms are
shown as anisotropic in the Atoms tab. Note that the “Plot?” button does not
produce this plot with obs & calc curves; it is intended only as a data
inspection tool.
Step 5. Doing a
combined neutron/x-ray PDF fit with PDFfit (optional)
If
you wish, you can add the available x-ray PDF to this project and use PDFfit to
fit both with a common Ni structure.
At
the bottom of the RMC tab for PDFfit, press the “Xray real space data” Select button. A file dialog will appear. Navigate to PDFfit-I/data and select Ni-xray.gr; the RMC tab will be
redrawn. Set the same flags as you used for the neutron G(r) entry. When done
the window should look like
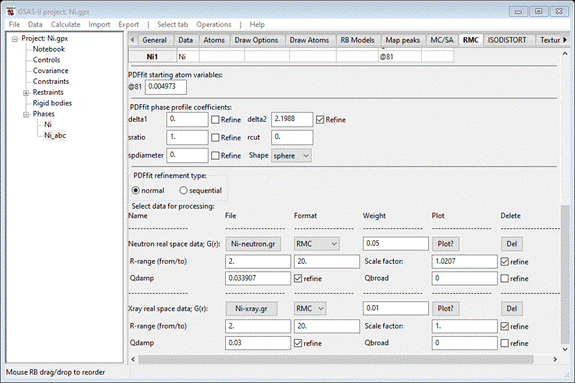
Next,
do Operations/Setup RMC and then do Operations/Execute. Again, the fit will proceed
very quickly; the console will show

The
Rw = 7.00% is very good. Press any key to get back to GSAS-II. After
accepting the results, the RNC window will be redrawn showing (at the bottom)
both data sets & their new parameters
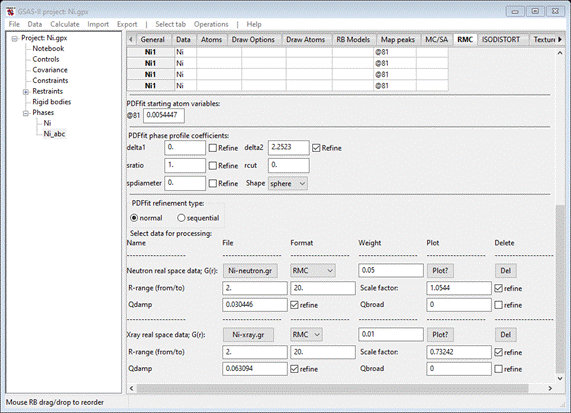
Do Operations/Plot; two plots will appear. One
is for the neutron data and the other for the x-ray data. For Ni they are
virtually identical; I show them here side-by-side. This can be done in GSAS-II
by dragging one plot tab to the right edge of the window; a light blue frame
will appear to show the screen has been split. Release the mouse button. You can
undo this by dragging the tab back to the main plot tab set.
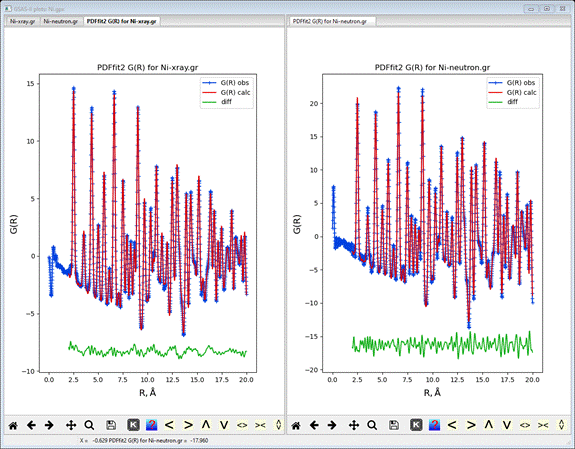
If
you examine the results file Ni_abc-PDFfit.res, you can find a short table near
the end
Refinement
parameters:
1: 1.05439
(0.013) 2: 0.0304455
(0.0026) 3: 0.732419 (0.00073)
4: 0.0630938
(0.00011) 10: 2.25235 (0.015) 11: 3.53157 (1.9e-05)
81: 0.00544471
(6.9e-06)
The
values with esds are given for each refined parameter; the parameter numbers
are those used in the “@N” symbolism used by PDFfit. These have been
automatically assigned by GSAS-II according to the type of parameter to avoid
unintended conflicts. The first two are scale & Qdamp for the neutron data
set and the next two are those for the x-ray data set. Numbers 1-9 are reserved
for these types of data parameters. #10 is for the delta2 phase parameter and
#11 is the Ni lattice parameter. Numbers 10-19 are reserved for phase &
lattice parameters. Although none are used in this example, atom coordinates
use parameter numbers in the range 20-79. Finally, #81 is the Ni Uiso term:
thermal motion parameter numbers are 81 or greater. The next tutorial will give
more complex uses of these parameter codes.
This
completes this introductory tutorial for PDFfit in GSAS-II; you can save your
project if you wish.